Biomedical and Health Sciences

The dataset contains 877 records with 67 variables, documenting various COVID-19-related indicators in Indonesia. The data spans daily case statistics, including new cases, cumulative cases, recoveries, and fatalities. It also tracks government response measures, such as school and workplace closures, public event restrictions, travel controls, and mask mandates. Additionally, it includes vaccination statistics, government response indices, and mobility changes in different sectors (retail, workplaces, and residential areas).
- Categories:
 184 Views
184 Views
The dataset contains 877 records with 67 variables, documenting various COVID-19-related indicators in Indonesia. The data spans daily case statistics, including new cases, cumulative cases, recoveries, and fatalities. It also tracks government response measures, such as school and workplace closures, public event restrictions, travel controls, and mask mandates. Additionally, it includes vaccination statistics, government response indices, and mobility changes in different sectors (retail, workplaces, and residential areas).
- Categories:
101 Views
This dataset comprises 2 million synthetic samples generated using the Variational Autoencoder-Generative Adversarial Network (VAE-GAN) technique. The dataset is designed to facilitate cardiovascular disease prediction through various demographic, physical, and health-related attributes. It contains essential physiological and behavioral indicators that contribute to cardiovascular health.
Dataset Description The dataset consists of the following features:
- Categories:
277 Views
This dataset comprises 2 million synthetic samples generated using the Variational Autoencoder-Generative Adversarial Network (VAE-GAN) technique. The dataset is designed to facilitate cardiovascular disease prediction through various demographic, physical, and health-related attributes. It contains essential physiological and behavioral indicators that contribute to cardiovascular health.
Dataset Description The dataset consists of the following features:
- Categories:
436 Views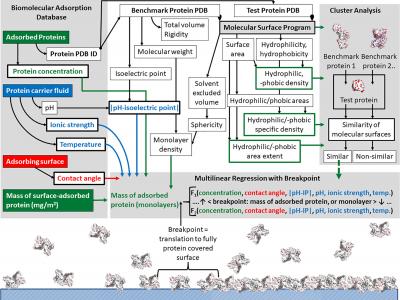
The Biomolecular Adsorption Database, BAD, is an archive of the data regarding protein adsorption on flat solid surfaces, as reported in peer-reviewed literature.
- Categories:
91 Views
A collection of Python pickles objects containing a Pandas DataFrame. Each Dataframe corresponds to the postprocessed firing rate (fr) in Hz and mean amplitude of the spikes (AMP) in microV/s of the vagus nerve recordings obtained from 12 adult female Sprague-Dawley rats. Additionally, the blood-glucose level in mg/dL is included. The fr and AMP signals have 0.1 miliseconds of resolution, whereas the glucose level was measured approximately every 5 minutes. Temporal variations are due to experimental factors. The number of available glucose samples changes across recordings.
- Categories:
42 Views
The dataset consists of two primary files: dataset.json and analysis_script.ipynb. The dataset.json file contains structured records of AI-assisted psychological therapy sessions, including emotion recognition, NLP techniques, cognitive behavioral therapy (CBT) patterns, hypnotherapy data, user feedback, and therapy outcomes. The analysis_script.ipynb Jupyter Notebook provides data preprocessing, visualization, and statistical analysis of therapy session outcomes.
- Categories:
1156 Views
The medical biometric dataset comprises 10,000 records collected across 23 patients spanning different demographics, biometric profiles, and temporal variations between 2022 and 2023. It is accumulated from various hospitals, digital health records, and biometric-enabled healthcare security systems.
- Categories:
204 Views
This dataset is designed for research on 2D Multi-frequency Electrical Impedance Tomography (mfEIT). It includes:
- Categories:
60 Views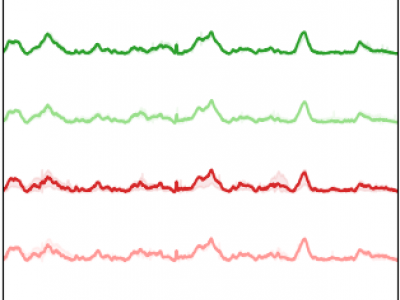
This dataset is from our paper "Bridging Lab-to-Clinic: Microbiological Screening via Swin-Ultra Transformer with Transfer Learning", which aims to validate the extension of the lab-verified bacterial classification model to the gene-type screening of unseen pathogens in clinical settings.
- Categories:
97 Views