GDB-9-Ex_TD-DFT-PBE0-SUBSET-100

- Citation Author(s):
-
Kshitij Mehta
 (Oak Ridge National Laboratory )
Massimiliano Lupo Pasini
(Oak Ridge National Laboratory )
Stephan Irle
(Oak Ridge National Laboratory )
Pilsun Yoo (Oak Ridge National Laboratory )Dmitry Ganyushin (Oak Ridge National Laboratory )
(Oak Ridge National Laboratory )
Massimiliano Lupo Pasini
(Oak Ridge National Laboratory )
Stephan Irle
(Oak Ridge National Laboratory )
Pilsun Yoo (Oak Ridge National Laboratory )Dmitry Ganyushin (Oak Ridge National Laboratory ) - Submitted by:
- Kshitij Mehta
- Last updated:
- DOI:
- 10.21227/8v6h-9e10
- Data Format:
- Research Article Link:
 51 views
51 views
- Categories:
- Keywords:
Abstract
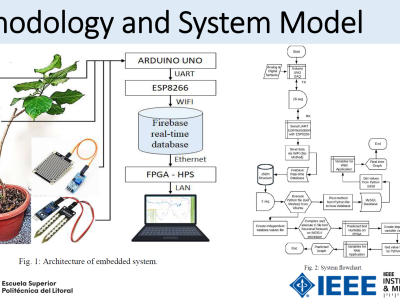
This is a subset of the original GDB-9-Ex_TD-DFT-PBE0 dataset at https://doi.org/10.13139/OLCF/2318314. It consists of 100 randomly selected molecules from the original dataset that consists of 96,766 molecules. The dataset contains data-intensive quantum chemical electronic structure calculations for organic molecules of the GDB-9-Ex dataset. Calculations were performed using the Time Dependent Density Functional Theory (TDDFT) first principles method using the ORCA software. It provides UV-vis spectra calculations of molecules with a high level of accuracy. The optical spectra behavior was collected based on the optimized molecular geometries in the DFTB method with 3ob parameters. All calculations utilized the def2-TZVP basis sets with the auxiliary def2/J and def2-TZVP/C basis sets. The time-dependent density-functional theory (TDDFT) approach with the PBE0 exchange-correlation functional and ORCAs default integration grid was employed. For the excitation energy calculations, the lowest 50 excitation states were calculated.
Instructions:
The dataset consists of 100 directories, one for each of the 100 molecules in the dataset. Each directory contains two files:
geo_end.xyz: ASCII file containing optimized geometry of the molecule with cartesian coordinates.
orca.stdout: Standard ASCII output generated by the ORCA software containing the output of the TDDFT calculations.
Molecule directories are named ‘mol_num’ where num corresponds to the id of the molecule from GDB-9-Ex dataset. It does not possess any practical significance as the geometry of the molecule is provided in the included geo_end.xyz file.
Total dataset size: 12 MiB